











فنیل کتونوریا (PKU) – اختلال ژنتیکی است که در آن آنزیم فنیل آلانین هیدروکسیلاز از بین رفته و یا به شدت دچار کمبود شده است، فنیل آلانین در تمامی غذاهای حاوی پروتئین (مانند گوشت، محصولات لبنی، ماهی و حبوبات) و در شیرین کننده های مصنوعی مثل آسپارتام شایع است. اجتناب از غذاهایی که حاوی فنیل آلانین، که به معنی یک رژیم غذایی کم پروتئین سخت، مانع از تجمع فنیل آلانین و آسیب های حاصل که در درجه اول برسیستم عصبی تاثیر می گذارد.نوعی بیماری ژنتیکی-متابولیکی اتوزومال (وابسته به کورموزمهای غیر جنسی) است؛ این اختلال حاصل جهش ژنی است که ساختیک آنزیم (فنیل کتونوریا هیدروکسیلاز) را در کنترل دارد.
این آنزیم اسید آمینه فنیل آلانین را (که به فراوانی در شیر مادر یافت می شود) به اسید آمینه ی دیگری (تیروزین) تبدیل می کند.
در نوزاد PKU بنا به دلایل گفته شده این آنزیم (فنیل کتونوریا هیدروکسیلاز) در کبد وجود ندارد. در نتیجه در بدو تولد و شیرخوارگی، اسید آمینه ی فنیل آلانین در بدن فرد تجزیه نمی گردد و مقادیر زیادی از این ماده ی شیمیایی خطرناک در بدن فرد انباشته می شود. انباشت فنیل آلانین در خون و سایر مایعات بدن موجب صدمات و آسیبهای جبران ناپذیری به بافتهای بدن بویژه مغز می گردد.
کودکان فنیل کتونوریا در ابتدای تولد ظاهراً شبیه به کودکان سالم هستند و هیچ علائمی را در بدو تولد نشان نمی دهند، اما اثرات این بیماری خیلی زود با صدماتی جبران ناپذیرش هویدا می گردد. آن هنگام که دیگر خیلی دیر شده است و برای این کودکان هیچ کاری نمی شود انجام داد؛ کودک فنیل کتونوریا در اثر تجمع ماده سمی فنیل آلانین هر ماه حدود 4 نمره ازبهره هوشی IQ خود را از دست می دهد.
ماحصل اختلال فنیل کتونوریا عقب ماندگی غالباً شدید ذهنی و برخی ناهنجاریهای دیگر( بیش فعالی، اختلالات گفتاری، تشنج و…) است. که با بالا رفتن سن کودک آشکار می شوند. رِنج ابتلای این بیماری حدود 1 در 10000 است اما در ایران این نرخ حدود 1 در 8000 است.
شیوه انتقال :
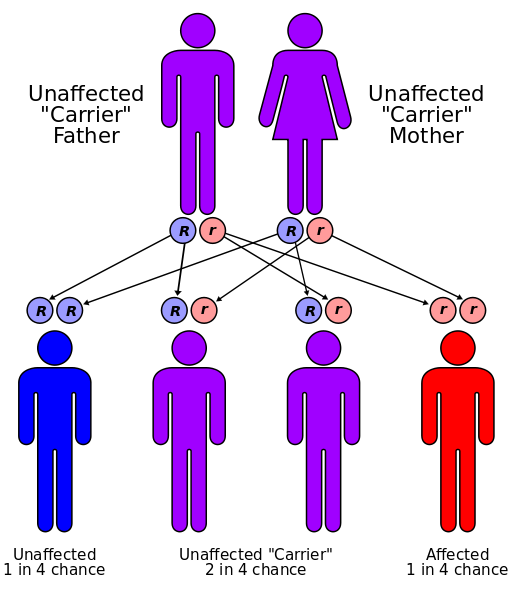
این بیماری به صورت اتوزومال مغلوب به ارث میرسد. ژن این بیماری بر روی کروموزم ۱۲ قرار گرفتهاست. چنانچه والدین هردو حامل این ژن باشند (که معمولاً در ازدواجهای خویشاوندی این احتمال بالاتر است) هر فرزند ، ٪۲۵ احتمال دارد که به فنیل کتونوریا مبتلا باشند. و این هشداری است به عزیزانی که قصد ازدواج فامیلی دارند.
تشخیص:
خوشبختانه امکان تشخیص این بیماری در بدو تولد وجود دارد. در کشورهای پیشرفته در بدو تولد با چند قطره خون که از پاشنه پای کودک گرفته میشود، میتوانند در حدود ۳۰ بیماری ژنتیکی از جمله پی کی یو را تشخیص دهند که اکثر این بیماریها قابل درمان یا کنترل هستند. در روز دوم یا سوم تولد با آزمایش تشخیصی که هم از طریق ادرار و هم از طریق خون میسر است میتوان بیماری را تشخیص داد. البته اگر از طریق خونگیری آزمایش انجام شود بهتر است.
در برحی از کشورها آزمایشات غربالگری فنیلکتونوریا برای همه ی نوزادان متولد شده انجام می گیرد. در ایران نیز در شهرهای بزرگ (تهران ، فارس….) اینگونه غربالگری انجام میگیرد. (28 دیماه ،روز غربالگری پی کی یو در ایران نامگذاری شده است)
البته با وجود آزمایشگاههای ژنتیکی مجهز، خوشبختانه اخیرا امکان تشخیص بیماری جنین در ماههای اول بارداری وجود دارد. در صورت تشخیص ابتلای جنین به بیماری، امکان سقط وی وجود دارد. همچنین با مشخص شدن نقص ژنتیکی، امکان مشاوره افراد فامیل بیمار که قصد دارند با یکدیگر ازدواج کنند، وجود خواهد داشت.
اگر یک زوج ناقل بیماری باشند، براساس اطلاعات کسب شده در مشاوره ژنتیک، میتوانند برای زندگی آینده خود تصمیم بگیرند.
البته ممکن است فردی ناقل باشد ولی خودش بیمار نباشد.
درمان (کنترل) :
خود این بیماری درمان قطعی ندارد اما هنگام تشخیص با رژیمهای غذایی خاص می توان تا حد زیادی از این عوارض جلوگیری کرد. همانگونه که گفته شد راه در مان این بیماری استفاده از شیرهای بدون فنیل آلانین(شیر لوفنالاک یا فنیل فری) است. به عبارتی با حذف فنیل آلانین در برنامه ی غذایی این افراد می توان از بروز صدمات یاد شده جلوگیری کرد. در صورت تشخیص زودرس و شروع استفاده از شیر مخصوص «فنیل کتونوری» از ابتدای نوزادی ، پیشآگهی خوب خواهد بود و کودک مبتلا میتواند با رعایت رژیم کتنرل کننده، از هوش طبیعی و رفتار مناسب برخوردار شود و با رعایت رژیم مخصوص و کنترل سطح فنیل آلانین خون، زندگی سالم و بی هیچ مشکلی را داشته باشد.. ولی تأخیر در درمان، به بروز عقبماندگی ذهنی، کوچکی دورسر و اختلالات رفتاری و تشنج منجر خواهد شد.
=============================================================
بطور مختصر:
ازدواجهای فامیلی ، عامل عمده ی شیوع این بیماری در جهان است. شاید خود شما هم بارها شنیده باشید که افرادی ازدواج فامیلی داشته و فرزندشان به عقب ماندگی ذهنی یا نوعی معلولیت دچار است. بنابراین پیشنهاد میشود درصورت تمایل به ازدواج فامیلی ، آزمایش و مشاوره ی ژنتیکی نیز انجام گیرد. اکثر افراد معلول ذهنی یا جسمی ، فرزندان کسانی هستند که ازدواج فامیلی داشته اند.افراد مبتلا به فنیل کتونوری، بالاترین محدودیت را در خوردن و آشامیدن دارند. آنها از خوردن نان،شیر،برنج،انواع لبنیات،گوشت،مرغ ، تخم مرغ، و ماهی،حبوبات،انواع تنقلات، بستنی،و… محرومند.
اگر کودکی را دیدید که در ماههای بدوی تولدش رفتاری عادی داشته ولی کم کم شروع به بیقراری و استفراغ شدید پس از خوردن شیر مادر و نیز تغییر رنک مو (تیره به روشن)و روشن شدن رنگ پوست و عدم رشد دور سر کرد، وظیفه ی شماست که سریعا به خانواده اش هشدار دهید که حتما از کودک، آزمایش ژنتیکی بعمل بیاورند. تاکید ما بر این است که هیچ ترسی از انجام این آزمایشات نداشته باشید. چون اگر کودک سالم باشد آسوده خاطر میشوید. و نیز اگر خدای ناخواسته کودک به بیماری ژنتیکی مبتلا باشد، از همان ابتدا باید تحت رژیم مخصوص غذایی قرار گیرد تا از زوال مغز و هوش جلوگیری شود و این باعث رشد طبیعی نوزاد میشود …
منبع-انجمن حمایت از بیماران فنیل کتونوریای اصفهان
سلام خدا قوت
میخواستم بپرسم با ژن درمانی نمیشه این بیماری رو درمان کرد؟